
Needleman–Wunsch
Global alignment algorithms are used to align two globally similar sequences. Commonly global alignment is used to identify first whether two protein, DNA or RNA sequences are homologous, and if so which amino acid residues (in proteins) or nucleotide sites (in DNA/RNA) are homologous.
For example, we might be interested in an important regulatory protein in Medicago truncatula (a close relative of alfalfa) called SUNN. When we perform a global alignment of SUNN against the protein CLV1 from Arabidopsis thaliana (one of the most well studied model plants), we see that despite more than 100 million years of separation between the two species the two proteins remain very similar. Very few gaps are inferred and most residues can be aligned.

We can see that the nine residues DVAIKRLV from column 719 to column 727 are
perfectly conserved. Experimentally the central lysine (K) in this region has
been shown to be essential for CLV1’s catalytic function in Arabidopsis
(Stone et al. 1998).
Based on the perfect sequence identity, it is reasonable to hypothesize that
these residues are also required for SUNN’s catalytic function.
Given an alignment of two proteins, a substitution matrix and some gap penalty system, it is trivial to calculate an alignment score. Many substitution matrices have been developed from curated empirical data sets, and for a variety of different purposes. For example the BLOSUM50 substitution matrix below is intended to be used for analyzing more divergent proteins:
| A | R | N | D | C | Q | E | G | H | I | L | K | M | F | P | S | T | W | Y | V | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 5 | -2 | -1 | -2 | -1 | -1 | -1 | 0 | -2 | -1 | -2 | -1 | -1 | -3 | -1 | 1 | 0 | -3 | -2 | 0 |
| R | -2 | 7 | -1 | -2 | -4 | 1 | 0 | -3 | 0 | -4 | -3 | 3 | -2 | -3 | -3 | -1 | -1 | -3 | -1 | -3 |
| N | -1 | -1 | 7 | 2 | -2 | 0 | 0 | 0 | 1 | -3 | -4 | 0 | -2 | -4 | -2 | 1 | 0 | -4 | -2 | -3 |
| D | -2 | -2 | 2 | 8 | -4 | 0 | 2 | -1 | -1 | -4 | -4 | -1 | -4 | -5 | -1 | 0 | -1 | -5 | -3 | -4 |
| C | -1 | -4 | -2 | -4 | 13 | -3 | -3 | -3 | -3 | -2 | -2 | -3 | -2 | -2 | -4 | -1 | -1 | -5 | -3 | -1 |
| Q | -1 | 1 | 0 | 0 | -3 | 7 | 2 | -2 | 1 | -3 | -2 | 2 | 0 | -4 | -1 | 0 | -1 | -1 | -1 | -3 |
| E | -1 | 0 | 0 | 2 | -3 | 2 | 6 | -3 | 0 | -4 | -3 | 1 | -2 | -3 | -1 | -1 | -1 | -3 | -2 | -3 |
| G | 0 | -3 | 0 | -1 | -3 | -2 | -3 | 8 | -2 | -4 | -4 | -2 | -3 | -4 | -2 | 0 | -2 | -3 | -3 | -4 |
| H | -2 | 0 | 1 | -1 | -3 | 1 | 0 | -2 | 10 | -4 | -3 | 0 | -1 | -1 | -2 | -1 | -2 | -3 | 2 | -4 |
| I | -1 | -4 | -3 | -4 | -2 | -3 | -4 | -4 | -4 | 5 | 2 | -3 | 2 | 0 | -3 | -3 | -1 | -3 | -1 | 4 |
| L | -2 | -3 | -4 | -4 | -2 | -2 | -3 | -4 | -3 | 2 | 5 | -3 | 3 | 1 | -4 | -3 | -1 | -2 | -1 | 1 |
| K | -1 | 3 | 0 | -1 | -3 | 2 | 1 | -2 | 0 | -3 | -3 | 6 | -2 | -4 | -1 | 0 | -1 | -3 | -2 | -3 |
| M | -1 | -2 | -2 | -4 | -2 | 0 | -2 | -3 | -1 | 2 | 3 | -2 | 7 | 0 | -3 | -2 | -1 | -1 | 0 | 1 |
| F | -3 | -3 | -4 | -5 | -2 | -4 | -3 | -4 | -1 | 0 | 1 | -4 | 0 | 8 | -4 | -3 | -2 | 1 | 4 | -1 |
| P | -1 | -3 | -2 | -1 | -4 | -1 | -1 | -2 | -2 | -3 | -4 | -1 | -3 | -4 | 10 | -1 | -1 | -4 | -3 | -3 |
| S | 1 | -1 | 1 | 0 | -1 | 0 | -1 | 0 | -1 | -3 | -3 | 0 | -2 | -3 | -1 | 5 | 2 | -4 | -2 | -2 |
| T | 0 | -1 | 0 | -1 | -1 | -1 | -1 | -2 | -2 | -1 | -1 | -1 | -1 | -2 | -1 | 2 | 5 | -3 | -2 | 0 |
| W | -3 | -3 | -4 | -5 | -5 | -1 | -3 | -3 | -3 | -3 | -2 | -3 | -1 | 1 | -4 | -4 | -3 | 15 | 2 | -3 |
| Y | -2 | -1 | -2 | -3 | -3 | -1 | -2 | -3 | 2 | -1 | -1 | -2 | 0 | 4 | -3 | -2 | -2 | 2 | 8 | -1 |
| V | 0 | -3 | -3 | -4 | -1 | -3 | -3 | -4 | -4 | 4 | 1 | -3 | 1 | -1 | -3 | -2 | 0 | -3 | -1 | 5 |
The scale of the above matrix is in third-bits, or log2(odds ratio) × 3. Another common scale is half-bits, or log2(odds ratio) × 2. Let’s calculate the alignment score for the first ten columns given our CLV1/SUNN alignment, the above BLOSUM50 substitution matrix, and a linear gap penalty of -8:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| CLV1 | M | A | M | R | L | L | K | T | H | L |
| SUNN | M | – | – | K | N | I | T | C | Y | L |
| Score | 7 | -8 | -8 | 3 | -4 | 2 | -1 | -1 | 2 | 5 |
When two residues are aligned we use the score from the substitution matrix. When a gap is present in either sequence we instead use the gap penalty of -8. Summing the scores together, we get a total score of -3 for the first ten columns.
Could we evaluate every possible global alignment of two sequences to find the alignment or alignments which optimize this score? For two sequences of length 2, there are 13 total alignments. This includes the gapless alignment, plus the following 12 alignments:
AB- -AB AB- AB-- A-B- A--B
A-B A-B -AB --AB -A-B -AB-
A-B A-B -AB --AB -A-B -AB-
AB- -AB AB- AB-- A-B- A--B
A general formula for calculating the number of global alignments for two sequences of of lengths m and n was reported by Torres et al.:
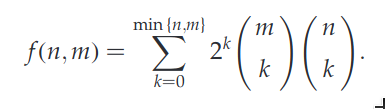
The number of sequences, as calculated using this forumla, quickly becomes unmanageable as n increases. There are more than one trillion possible global alignments for two sequences of length 17 each:
| n | # global alignments | N–W |
|---|---|---|
| 1 | 3 | 1 |
| 2 | 13 | 4 |
| 3 | 63 | 9 |
| 4 | 321 | 16 |
| 5 | 1,683 | 25 |
| 6 | 8,989 | 36 |
| 7 | 48,639 | 49 |
| 8 | 265,729 | 64 |
| 9 | 1,462,563 | 81 |
| 10 | 8,097,453 | 100 |
| 11 | 45,046,719 | 121 |
| 12 | 251,595,969 | 144 |
| 13 | 1,409,933,619 | 169 |
| 14 | 7,923,848,253 | 196 |
| 15 | 44,642,381,823 | 225 |
| 16 | 252,055,236,609 | 256 |
| 17 | 1,425,834,724,419 | 289 |
Given that proteins can be hundreds or thousands of amino acids in length, clearly we need smarter algorithms for global alignment. The Needleman–Wunsch algorithm (Needleman and Wunsch 1970) is a dynamic programming method to solve the global alignment problem with only n2 calculations (the N–W column in the table above, or if the two sequences are of different lengths n and m, nm calculations).
While the exact definition is contentious, dynamic programming basically works by storing intermediate results for subproblems which otherwise would have to be recomputed many times. Not all intermediate results must be stored if we are only interested in an optimal solution, as in the case of global alignment.
In the Needleman–Wunsch algorithm, the intermediate results are the optimal
alignment scores when the two sequences are truncated after two corresponding
sites or residues. For example, let’s align the first ten residues of
CLV1 X=MAMRLLKTHL with the first eight residues of SUNN Y=MKNITCYL.
First, draw up a table with n + 1 columns and m + 1 rows. You can also add an extra row at the top and column at the left to refer to the amino acid residues. This table is known as an F-matrix.
Write a zero in the top left cell, and add gap penalties to the first row and column. In this case we will use a linear gap penalty of -8, so simply decrement each cell by 8. Draw arrows (we can call them pointers in computer science lingo) leading back to the top left cell.
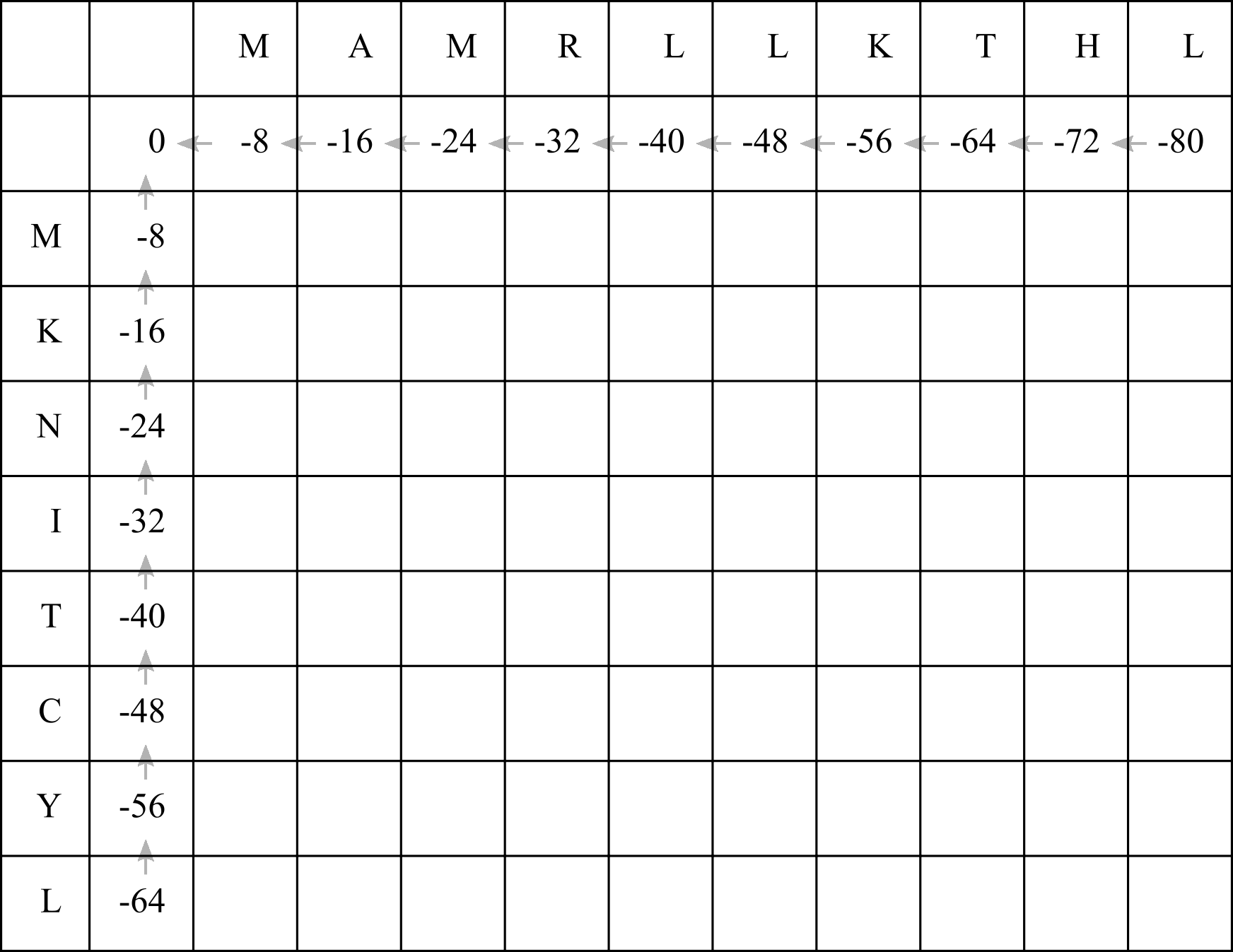
Now we will fill in the F-matrix. To fill in each cell we need to refer to the cell immediately above, to the left, and diagonally up and left. So we will begin with the first empty cell in the top left.
For every cell we fill in, we must evaluate three options:
- Use the score for the cell above and add a gap penalty
- Use the score for the cell to the left and add a gap penalty
- Add a score from the substitution matrix to the score for the cell diagonally up and left
For the first empty cell, we can add -8 to the cell above (the first option) or to the left (the second option). Both those cells have a score of -8, so in either case the final score will be -16. If we choose to align the two methionine (M) residues, the BLOSUM50 matrix gives us a score of 7. So adding 7 to the diagonal cell’s score of 0 gives us 7.
The best option is whatever gives us the highest score. We will therefore choose option 3, fill in the score of 7, and draw a diagonal arrow:
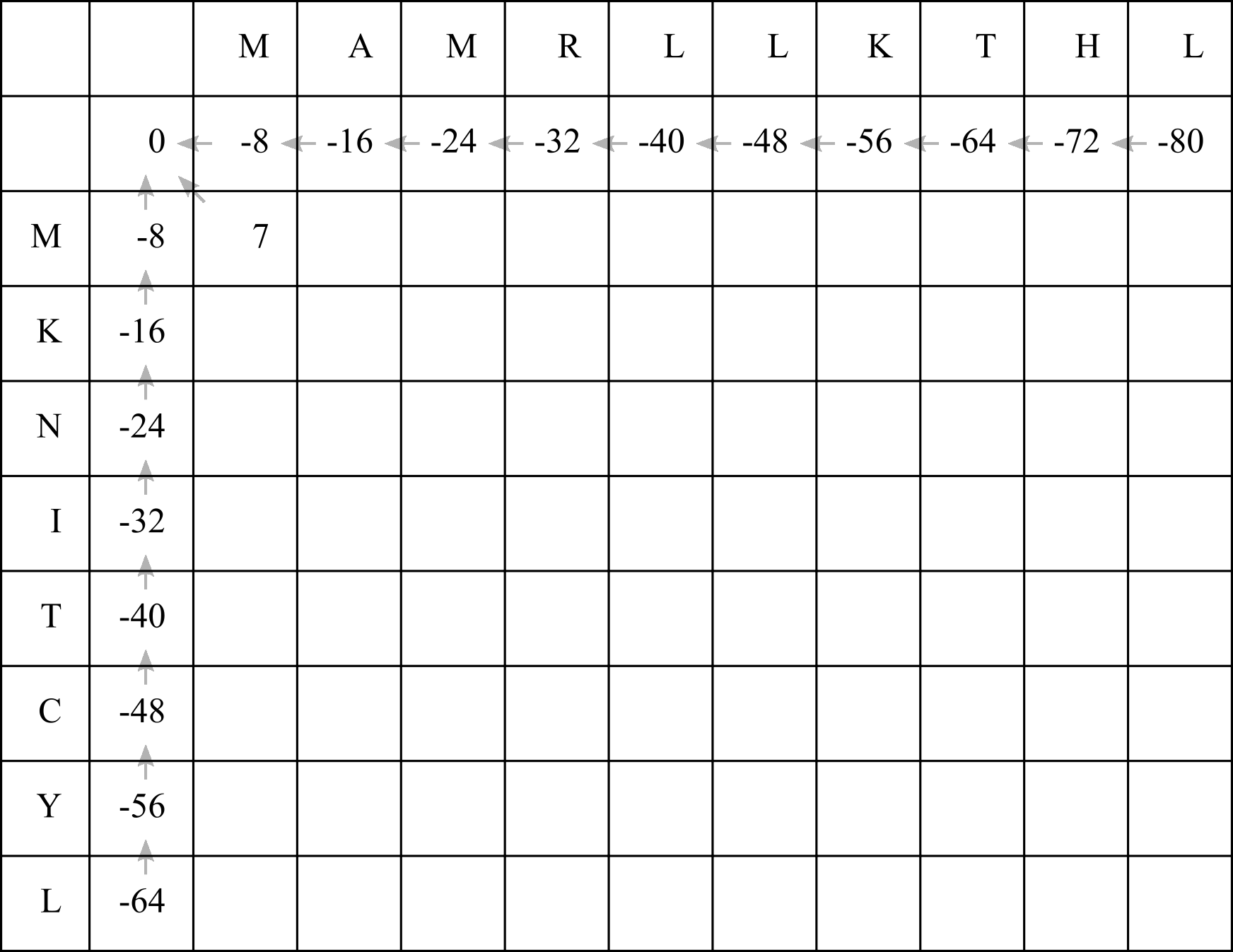
Now we can fill in the next cell. In this case the scores for each option will be:
- -16 + -8 = -24 (add a gap penalty to the above cell’s score)
- 7 + -8 = -1 (add a gap penalty to the left cell’s score)
- -8 + -1 = -9 (add the BLOSUM50 score for aligning M with A to the diagonal cell’s score)
Clearly for the second cell the best option is actually 2! Fill in a score of -1, and add a left arrow:
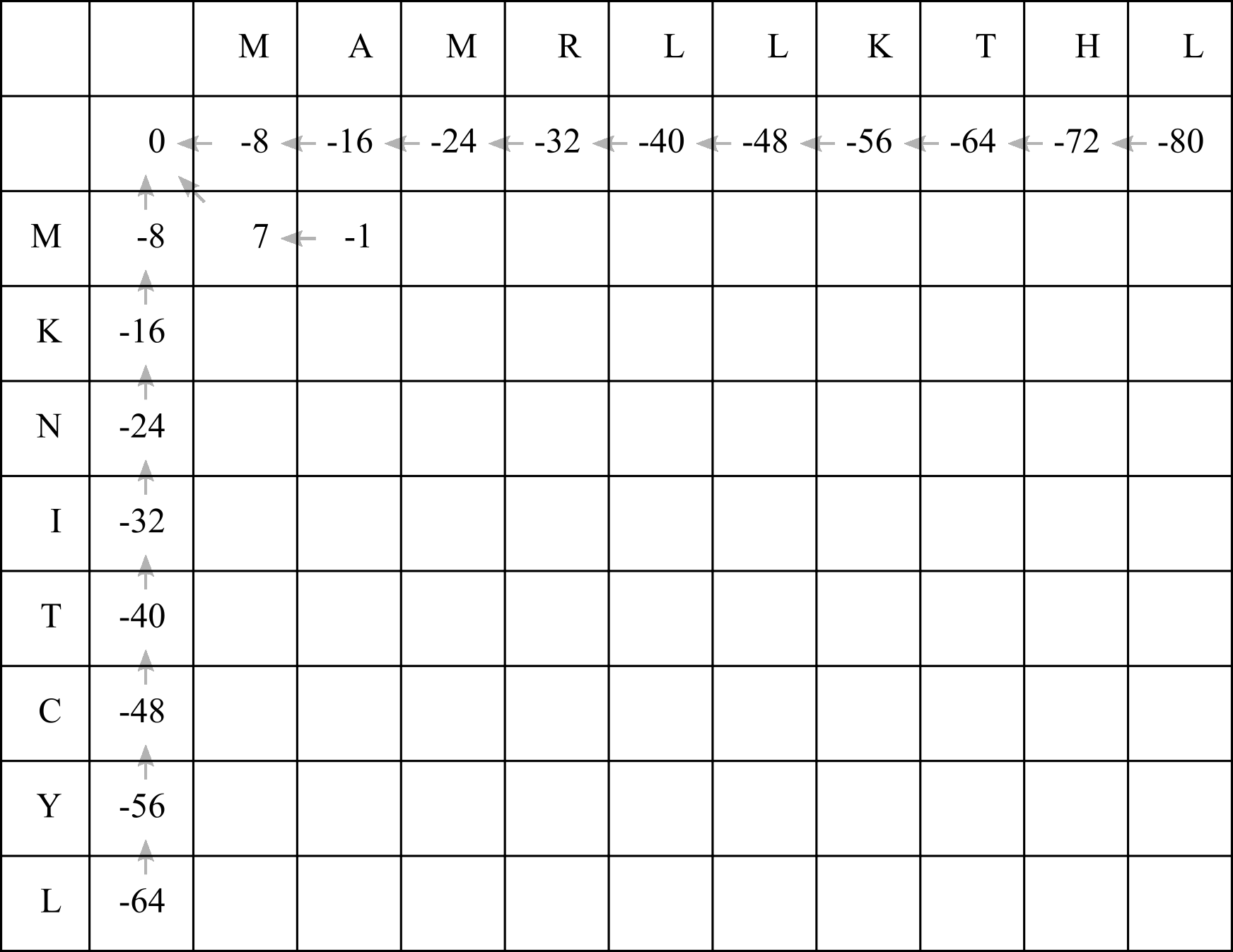
When filling in the F-matrix, it is possible that multiple options (up, left, diagonal) will produce the same highest score. In that case, you should add multiple arrows leading out of the table cell.
For the third cell, adding a gap penalty to the left cell’s score (-1 + -8) or aligning the two M residues and adding the BLOSUM50 score to the diagonal cell’s score (-16 + 7) both result in an equally high score of -9. An up arrow would have a much lower score of -24 + -8 = -32, so in this case draw arrows leading left and leading diagonally.
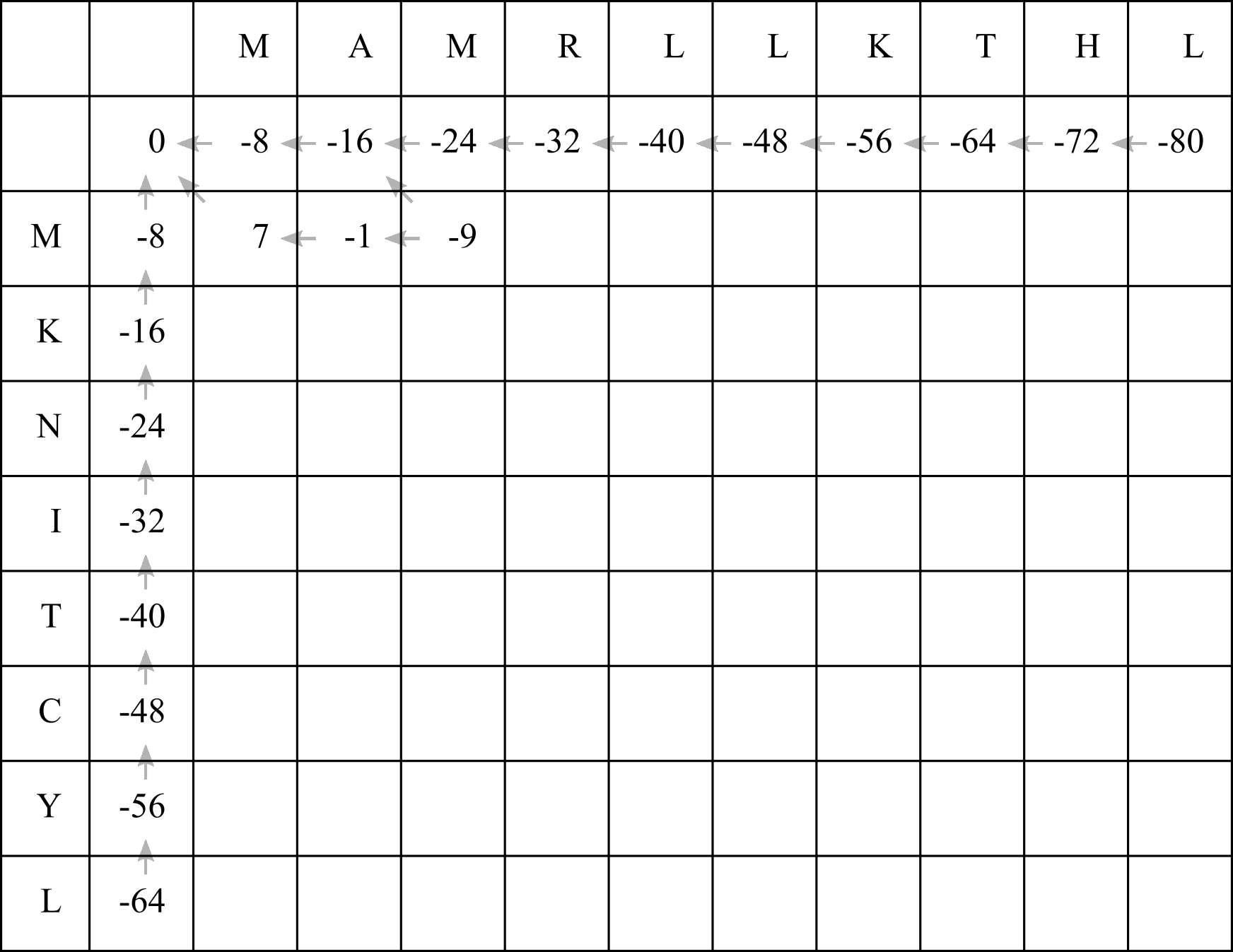
Now fill in the rest of the matrix:
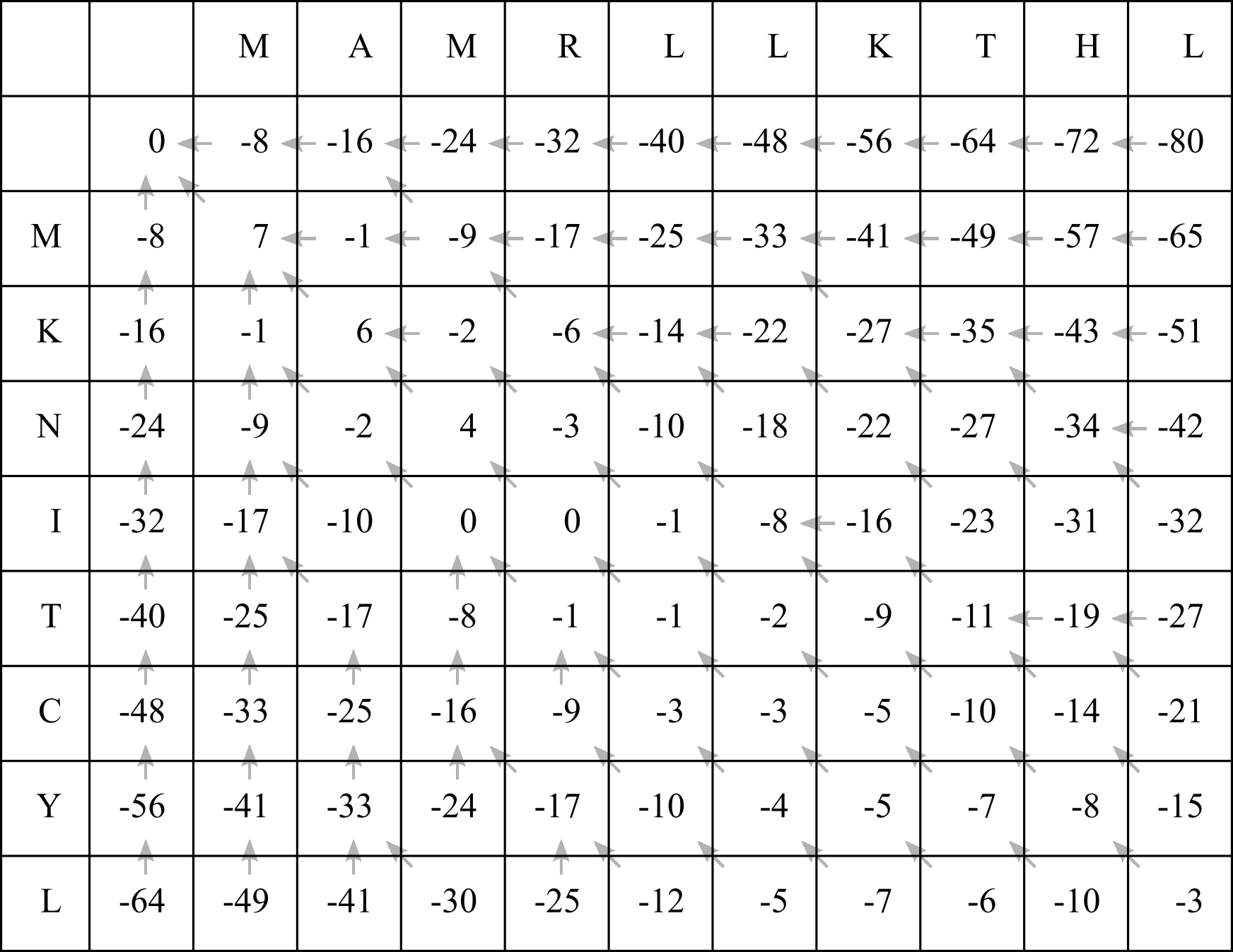
Interestingly, the score at any cell represents the highest possible alignment score if we had truncated the sequences after the corresponding X and Y sites. The score in the bottom right cell therefore represents the highest possible score for the entire two sequences being aligned.
To complete the alignment, we proceed from the end of the alignment (the C-terminus or 3’ end) to the beginning (N-terminus or 5’ end), tracing the arrows back through the F-matrix, and ALWAYS beginning in the bottom right cell. Again as for filling in the table, we follow some simple rules to generate the alignment.
Up arrow: Align the Y sequence residue/site with a gap
Left arrow: Align the X sequence residue/site with a gap
Diagonal arrow: Align both the X and the Y sequence residues/sites
In the CLV1/SUNN example, there are at least seven diagonal arrows beginning in the bottom right corner cell:
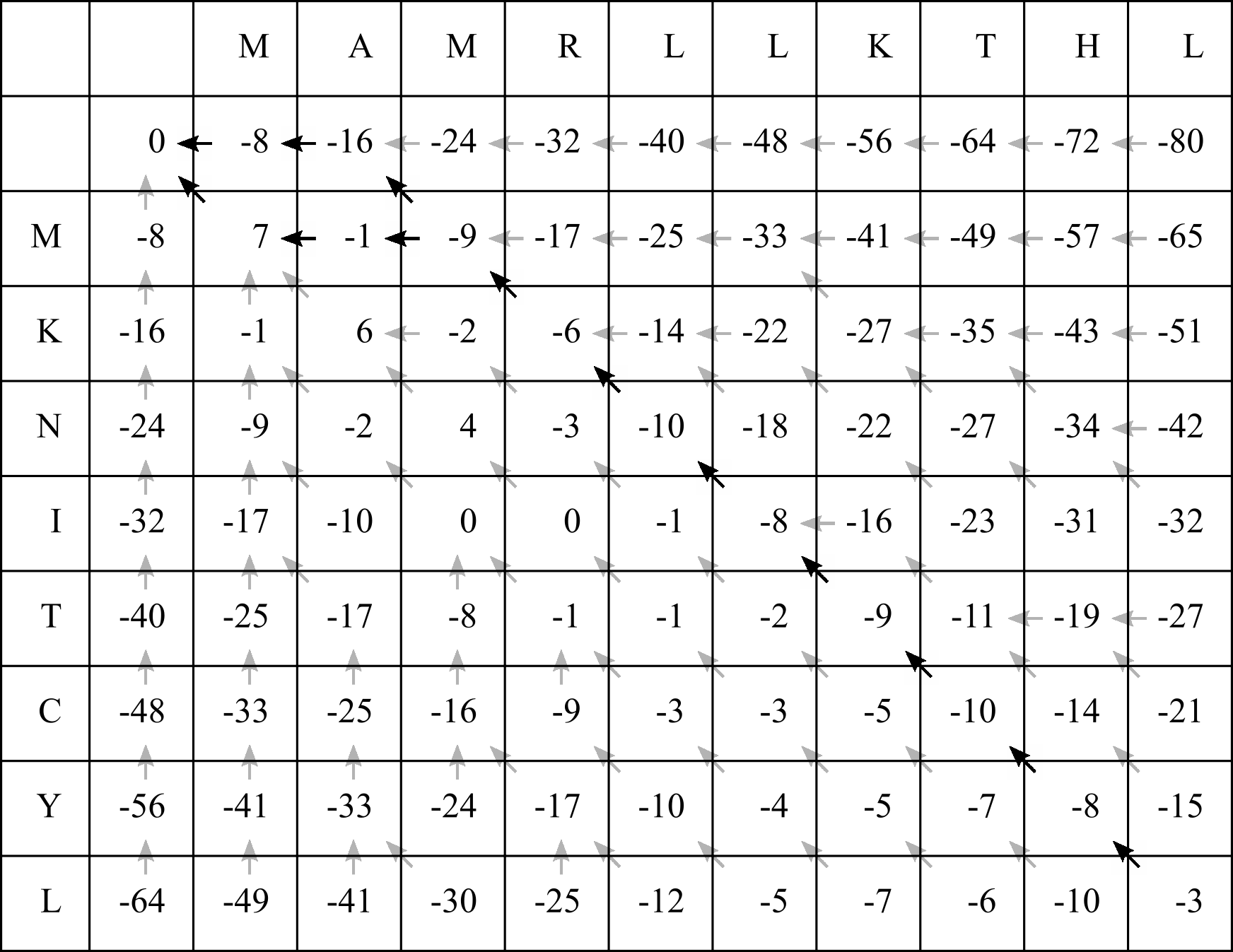
This means we will align the first seven residues (FROM THE RIGHT HAND SIDE) of both sequences to match the seven diagonal arrows:
RLLKTHL
KNITCYL
We now face a fork, and can choose left or diagonal. When reconstructing the alignment, you can choose the arrow to follow at random to identify a single optimal alignment. A forking recursive algorithm can also be used to identify all optimal alignments.
Let’s choose the diagonal arrow to align the next residue from both sequences, and then align the last two X residues with gaps to match the two left arrows:
MAMRLLKTHL
--MKNITCYL
If we instead follow the left path in the fork, the final alignment will be:
MAMRLLKTHL
M--KNITCYL
Looking at the final alignments, it should be fairly obvious why they are equally good choices, and based on CLV1 and SUNN protein sequences alone without any additional information, we can’t pick the correct alignment with certainty. You now know how to optimally globally align two molecular sequences by hand!
I have written a Python program to generate optimal global alignments automatically. Please read through the code and the comments. Then save it to your computer and run it in Python (this will work in versions 2 or 3 of Python, provided you have numpy installed).
import numpy
# The alphabet defines the order of amino acids used for the score matrix
# so A is the first character, R is the second etc.
alphabet = "ARNDCQEGHILKMFPSTWYV"
# The BLOSUM50 matrix in third-bit units, or 3log2(odds ratio)
# We store it as a 2D array using the numpy package
blosum50 = numpy.array([
[ 5,-2,-1,-2,-1,-1,-1, 0,-2,-1,-2,-1,-1,-3,-1, 1, 0,-3,-2, 0],
[-2, 7,-1,-2,-4, 1, 0,-3, 0,-4,-3, 3,-2,-3,-3,-1,-1,-3,-1,-3],
[-1,-1, 7, 2,-2, 0, 0, 0, 1,-3,-4, 0,-2,-4,-2, 1, 0,-4,-2,-3],
[-2,-2, 2, 8,-4, 0, 2,-1,-1,-4,-4,-1,-4,-5,-1, 0,-1,-5,-3,-4],
[-1,-4,-2,-4,13,-3,-3,-3,-3,-2,-2,-3,-2,-2,-4,-1,-1,-5,-3,-1],
[-1, 1, 0, 0,-3, 7, 2,-2, 1,-3,-2, 2, 0,-4,-1, 0,-1,-1,-1,-3],
[-1, 0, 0, 2,-3, 2, 6,-3, 0,-4,-3, 1,-2,-3,-1,-1,-1,-3,-2,-3],
[ 0,-3, 0,-1,-3,-2,-3, 8,-2,-4,-4,-2,-3,-4,-2, 0,-2,-3,-3,-4],
[-2, 0, 1,-1,-3, 1, 0,-2,10,-4,-3, 0,-1,-1,-2,-1,-2,-3, 2,-4],
[-1,-4,-3,-4,-2,-3,-4,-4,-4, 5, 2,-3, 2, 0,-3,-3,-1,-3,-1, 4],
[-2,-3,-4,-4,-2,-2,-3,-4,-3, 2, 5,-3, 3, 1,-4,-3,-1,-2,-1, 1],
[-1, 3, 0,-1,-3, 2, 1,-2, 0,-3,-3, 6,-2,-4,-1, 0,-1,-3,-2,-3],
[-1,-2,-2,-4,-2, 0,-2,-3,-1, 2, 3,-2, 7, 0,-3,-2,-1,-1, 0, 1],
[-3,-3,-4,-5,-2,-4,-3,-4,-1, 0, 1,-4, 0, 8,-4,-3,-2, 1, 4,-1],
[-1,-3,-2,-1,-4,-1,-1,-2,-2,-3,-4,-1,-3,-4,10,-1,-1,-4,-3,-3],
[ 1,-1, 1, 0,-1, 0,-1, 0,-1,-3,-3, 0,-2,-3,-1, 5, 2,-4,-2,-2],
[ 0,-1, 0,-1,-1,-1,-1,-2,-2,-1,-1,-1,-1,-2,-1, 2, 5,-3,-2, 0],
[-3,-3,-4,-5,-5,-1,-3,-3,-3,-3,-2,-3,-1, 1,-4,-4,-3,15, 2,-3],
[-2,-1,-2,-3,-3,-1,-2,-3, 2,-1,-1,-2, 0, 4,-3,-2,-2, 2, 8,-1],
[ 0,-3,-3,-4,-1,-3,-3,-4,-4, 4, 1,-3, 1,-1,-3,-2, 0,-3,-1, 5],
])
# Specify a linear GAP penalty of -8
gap_penalty = -8
# Our X and Y sequences to align
x = "MAMRLLKTHL"
y = "MKNITCYL"
# Set up our F-matrix using a two dimensional score_matrix to record the
# intermediate values and a three dimensional pointer_matrix to record the
# arrows. The first and second axes of the score_matrix and pointer_matrix
# correspond to the X and Y sequences respectively. The third axis of the
# pointer_matrix records the presence (1) or absence (0) of arrows in the
# diagonal (align both), left (align X with gap), and up (align Y with gap) at
# indices 0, 1 and 2 respectively. Because with have the first row and first
# column filled in with gap penalties, the row and column indices
# corresponding to the X and Y residues will begin at 1.
score_matrix = numpy.zeros((len(x) + 1, len(y) + 1), dtype = int)
pointer_matrix = numpy.zeros((len(x) + 1, len(y) + 1, 3), dtype = int)
# Fill in first row and column with the linear gap penalties
for xi, xaa in enumerate(x):
score_matrix[xi + 1, 0] = gap_penalty * (xi + 1)
pointer_matrix[xi + 1, 0, 1] = 1
for yi, yaa in enumerate(y):
score_matrix[0, yi + 1] = gap_penalty * (yi + 1)
pointer_matrix[0, yi + 1, 2] = 1
# Fill in middle values starting, from left to right in the top row, then from
# left to right in the second row and so on. The yi and xi indices begin at 0.
for yi, yaa in enumerate(y):
for xi, xaa in enumerate(x):
# Get the index (position) of the X and Y amino acids in our alphabet
xaai = alphabet.index(xaa)
yaai = alphabet.index(yaa)
# These are the indices to the X and Y positions in the F-matrix,
# which begin at 1 because of the the first row and first column of
# gap penalties.
fxi = xi + 1
fyi = yi + 1
# Look up the score for the X and Y pair of residues in our BLOSUM50 matrix
residue_score = blosum50[xaai, yaai]
# Calculate the possible scores for aligning both residues...
no_gap_score = score_matrix[fxi - 1, fyi - 1] + residue_score # (fxi - 1, fyi - 1) is the diagonally up and left cell
# ...and for aligning the X residue with a gap...
x_gap_score = score_matrix[fxi - 1, fyi] + gap_penalty # (fxi - 1, fyi) is the cell to the left of the current cell
# ...and for aligning the Y residue with a gap.
y_gap_score = score_matrix[fxi, fyi - 1] + gap_penalty # (fxi, fyi - 1) is the cell above the current cell
# Identify the maximum score value from among the three options...
max_score = max(no_gap_score, x_gap_score, y_gap_score)
# ...and set the score of this F-matrix cell to that score.
score_matrix[fxi, fyi] = max_score
# Add arrows for any direction where choosing that direction
# will result in the maximum score for this F-matrix cell
if no_gap_score == max_score:
pointer_matrix[fxi, fyi, 0] = 1
if x_gap_score == max_score:
pointer_matrix[fxi, fyi, 1] = 1
if y_gap_score == max_score:
pointer_matrix[fxi, fyi, 2] = 1
# A recursive function to regenerate the optimal alignment from the F-matrix,
# randomly sampling if there is more than one optimal alignment
def recurse_score_matrix(xi, yi):
# Once the top left corner, return two empty sequences (an empty pairwise alignment)
if xi == yi == 0:
return "", ""
# Pick one of the outgoing arrows at random. Firt, generate a random
# permutation of the sequence [0, 1, 2] corresponding to diagonal, left
# and up arrows respectively.
random_pointer_order = numpy.random.permutation(3)
# Next, find the first number in that permutation where the arrow at that
# index of the pointer array is present.
for random_pointer_choice in random_pointer_order:
if pointer_matrix[xi, yi, random_pointer_choice] == 1:
break
# Align two residues, or align x residue with a gap, or y residue with a gap.
# xj, yj will refer to the indices of the
if random_pointer_choice == 0: # along both residues (diagonal arrow)
xj = xi - 1 # xj is the index of the next cell (one index left)
yj = yi - 1 # yj is the index of the next cell (one index up)
# Get the X and Y sequence characters corresponding to the current
# cell. Remember that the indices for sequence characters begin at 0
# for the sequence strings but begin at 1 in the F-matrix, so we
# subtract 1 to get the correct indices in the sequence strings while
# we are traversing the F-matrix using xi, yi.
xc = x[xi - 1]
yc = y[yi - 1]
elif random_pointer_choice == 1: # align X with gap
xj = xi - 1
yj = yi
xc = x[xi - 1]
yc = "-" # use a dash for the gap
else: # align Y with gap
xj = xi
yj = yi - 1
xc = "-"
yc = y[yi - 1]
# Add the two characters (the two residues, or a residue and a gap) to the
# partial alignment generated by recursing through the next cell (pointed
# to by the randomly chosen arrow), and return the new partial alignment.
partial_x, partial_y = recurse_score_matrix(xj, yj)
partial_x += xc
partial_y += yc
return partial_x, partial_y
# Call the recursive function, beginning at the bottom right hand corner. The
# coordinate of the bottom right cell is equal to the length of the X sequence
# and the length of the Y sequence
optimal_alignments = recurse_score_matrix(len(x), len(y))
# Numpy treats the first axis as rows, and the second axis as columns. But we
# are treating the axes as X and Y respectively, so if we print the score matrix,
# X0 will be the first row, X1 will be the second etc. To make the X sequence
# run side-to-side we therefore transpose the matrix before printing.
print(score_matrix.transpose())
# Print the optimal alignments in separate lines (so they are aligned in the
# output).
print("\n" + optimal_alignments[0] + "\n" + optimal_alignments[1])
For more information about Needleman–Wunsch, see chapter 2 of Biological sequence analysis by Durbin et al., and Wikipedia.
Update 13 October 2019: for an another perspective on sequence alignment, including global alignment, consult Chapter 5 of Bioinformatics Algorithms (2nd or 3rd Edition) by Compeau and Pevzner.