
Phylogenetic trees and data structures
Bifurcating trees have found enormous utility in biology. Depending on what the tips of a tree represent, they can be used as a model to help understand various systems, processes and relationships. For example, the tips of the tree could represent different species, different genes, different alleles of the same gene, cells from different healthy tissues, cancer cells, or virus genomes.
Species trees
When the tips of a tree are different species, the tree represents the evolutionary history and relationship between those species. Darwin famously considered the use of trees for this purpose in one of his notebooks.
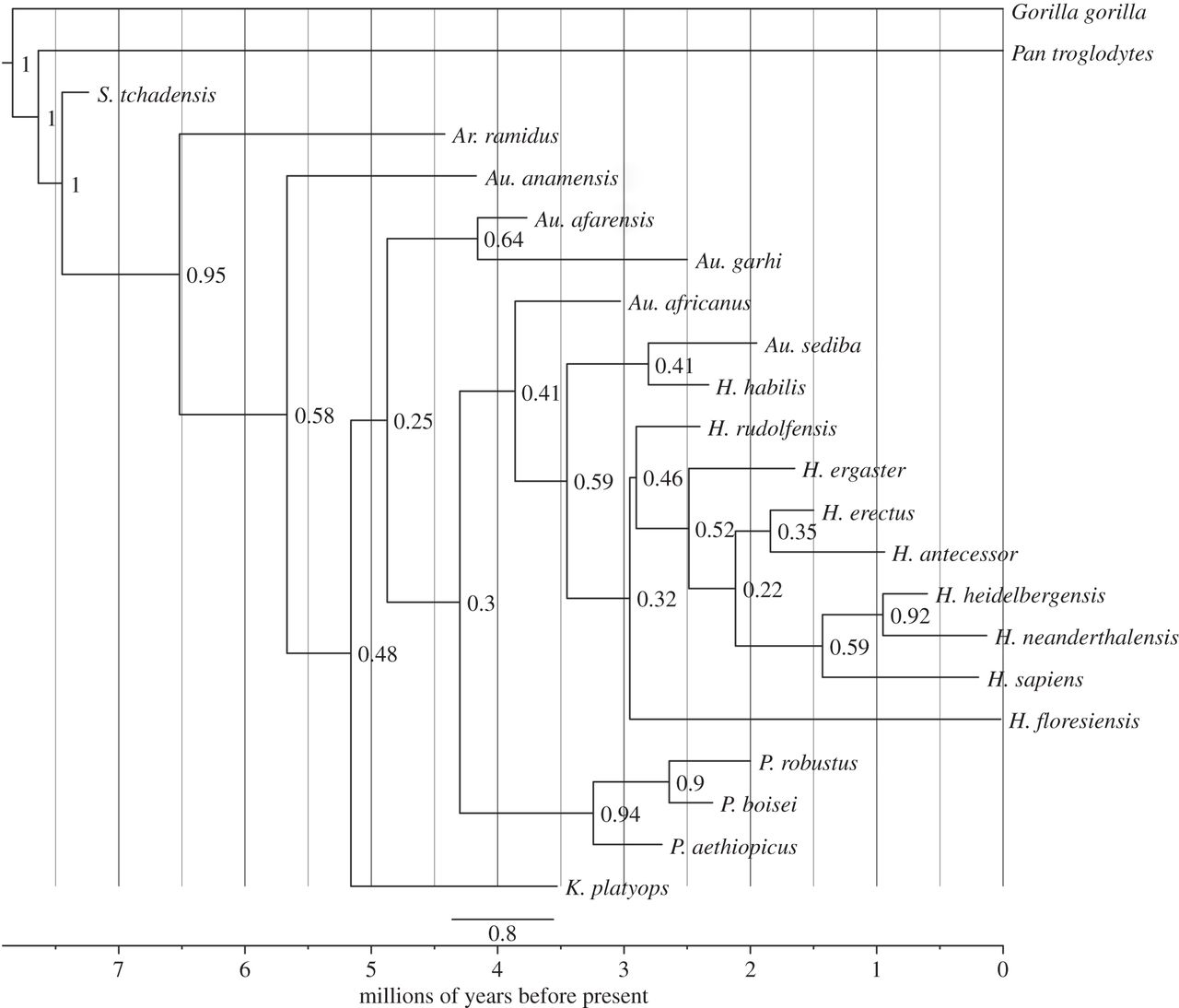
One example species tree would be this tree from Dembo et al. (2015) of humans and our close (but extinct) relatives:
Gene trees
Phylogenetic trees have revealed how color receptor proteins (opsins) have independently diversified in vertebrates and arthropods. See this tree from Pichaud et al. (1999):
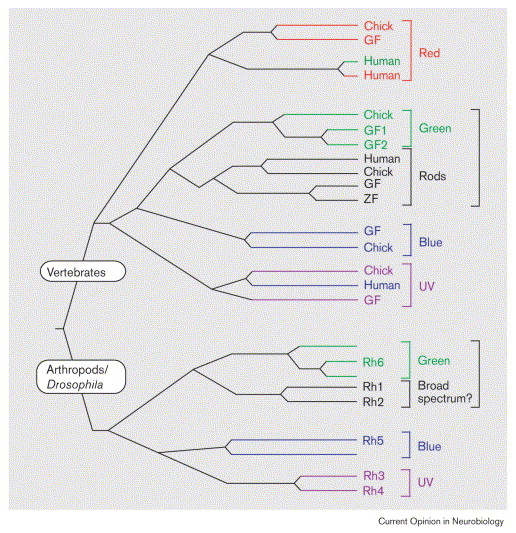
When two tips in a gene tree are related purely by speciation (as in the rod genes, where there is only a single rod gene for each species), they are called orthologs. They can also be described as different alleles of the same gene. Otherwise if the genes are different because of gene duplication, they are called paralogs. For example the human red and green color receptor genes are recently duplicated from a single common green ancestor.
Other trees
Trees can be used to model cell lineages, that is how cells differentiate all the way from embryonic stem cells to the skin, bone, red blood, neural, and all the other cells in our body.
Trees can be made of virus genomes. If the genomes are taken from multiple virions within a single host, the tree models the infection within that host. If the genomes are taken from hosts which directly infected each other, the tree models the transmission of the virus and is called a “transmission tree”. If the genomes are taken from a wider sample of hosts, the tree can be used to model an epidemic.
Tree data structures
Whatever is being represented, the trees can be stored in computer memory in one of three basic ways. Consider the following unrooted tree:
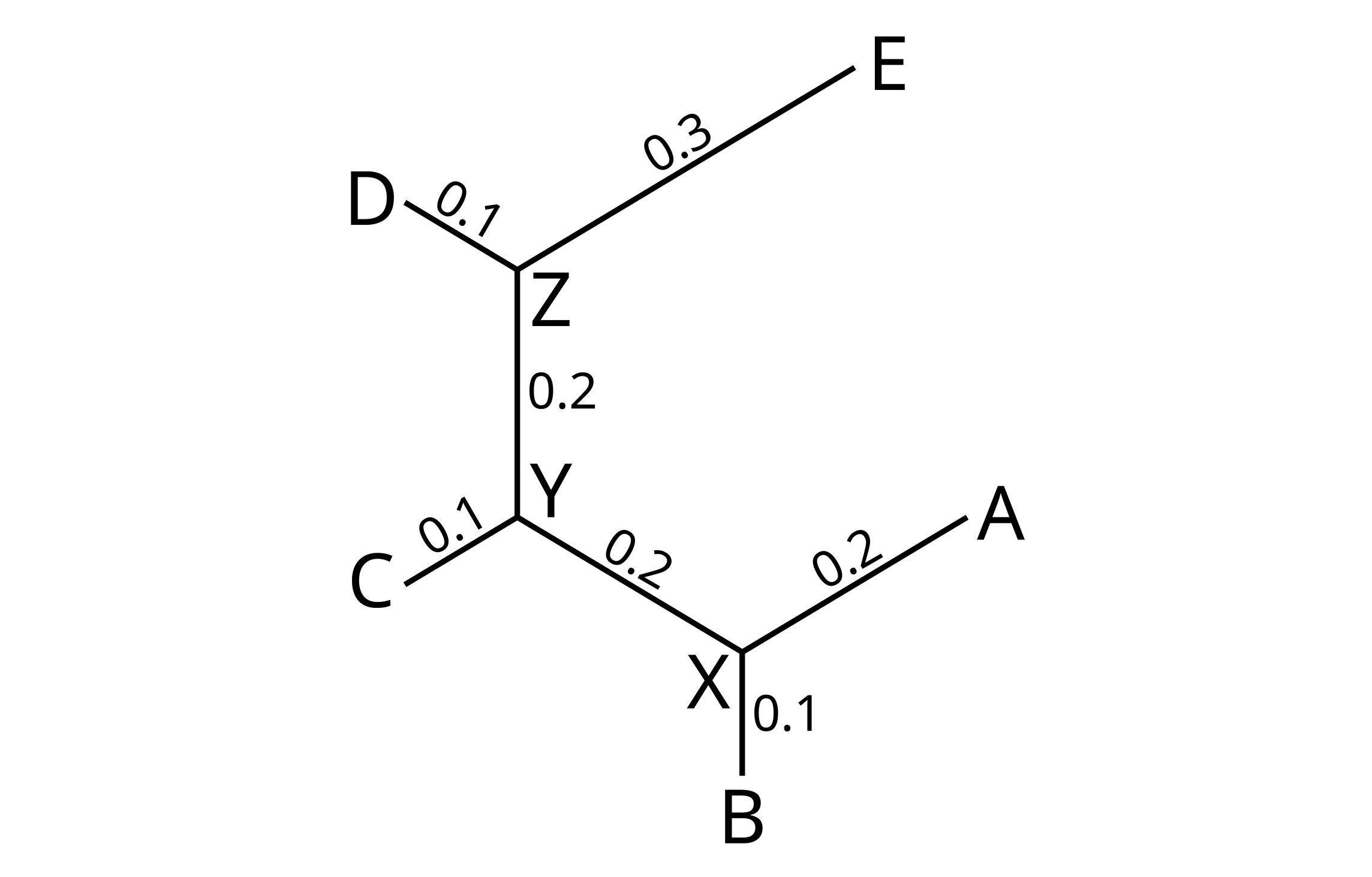
There are 5 external nodes (A through E), and 3 internal nodes (X, Y and Z). It can be stored as an adjacency matrix by leaving elements blank where no edge connects two nodes, and by including the branch length where there is such as edge:
| A | B | C | D | E | X | Y | Z | |
|---|---|---|---|---|---|---|---|---|
| A | 0.2 | |||||||
| B | 0.1 | |||||||
| C | 0.1 | |||||||
| D | 0.1 | |||||||
| E | 0.3 | |||||||
| X | 0.2 | 0.1 | 0.2 | |||||
| Y | 0.1 | 0.2 | 0.2 | |||||
| Z | 0.1 | 0.3 | 0.2 |
This data structure is seen rarely in phylogenetics. As you can see it is very space inefficient where most edges do not exist. The number of possible edges grows by n2, but the number of edges in an unrooted bifurcating tree grows by 2n - 3. Even in the above example where we double this number by storing edges symmetrically in the matrix, more and more elements will be empty as the number of tips in the tree increases.
More commonly uses is an adjacency list. In this data structure, for each node we store its edges using a data structure like a list, or set, or map. Those data structures can be stored as an array where the indices are mapped to the nodes in some way. In the following example, a map is used store the edges of a node using the adjacent nodes as the keys, and the branch lengths as the values:
| A | { X:0.2 } |
| B | { X:0.1 } |
| C | { Y:0.1 } |
| D | { Z:0.1 } |
| E | { Z:0.3 } |
| X | { A:0.2, B:0.1, Y:0.2 } |
| Y | { C:0.1, X:0.2, Z:0.2 } |
| Z | { D:0.1, E:0.3, Y:0.2 } |
Adjacency lists are a time and space efficient data structure for storing phylogenetic trees. But from a programming perspective, it can be easier to work with purely object-oriented trees, where each node is an instance (object) of some kind of node class. Edges can themselves be objects, or nodes can directly store their local relationships as pointers to each other. The ETE Toolkit, a phylogenetic library for Python, uses this kind of data structure.